Фенилкетонурия, что это за заболевание, симптомы у новорожденных, этиология, код по МКБ 10 у детей
Опубликовано: 01.09.2018
Наследственные заболевания возникают при патологическом изменении генетического аппарата. Часть подобных нарушений выявляется преимущественно после рождения ребенка, к их числу относят и фенилкетонурию.
Особенностью этого заболевания является то, что при своевременном его выявлении и соблюдении лечебного питания люди живут полноценной жизнью.
В противном случае накапливаются вредные (токсические) для организма соединения, вызывающие тяжелые функциональные нарушения со стороны внутренних органов, в том числе и ЦНС.
Описание патологии
Развитие фенилкетонурии (ФКУ) связано с неспособностью организма человека с дефектным геном расщеплять поступающую вместе с пищей, богатой на белки, аминокислоту под названием фенилаланин.
В итоге кислота и элементы ее распада скапливаются в тканях и биожидкостях, превращаются в токсические соединения, под воздействием которых происходит глубокое и в большинстве случаев необратимое поражение органов ЦНС.
Отсутствие своевременного адекватного лечения становится причиной задержки развития интеллекта вплоть до олигофрении.
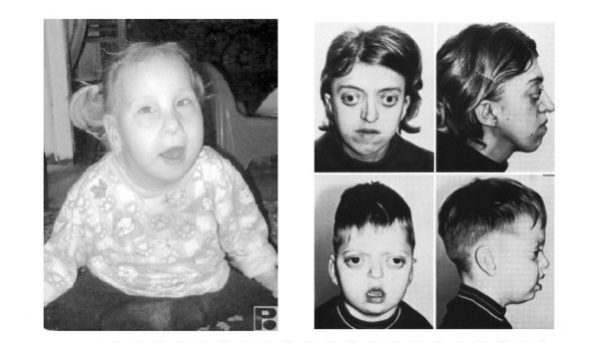
Манифестирует Фенилкетонурия обычно у больных малышей в первые 3-6 месяцев после рождения.
Выявление заболевания до появления первых клинических проявлений и соблюдение в дальнейшем эллиминационной диеты исключает развитие тяжелых, необратимых расстройств нервной системы у детей и взрослых, то есть опасности для физического, психоэмоционального и умственного здоровья нет.
Фенилкетонурия в среднем диагностируется у 1 новорожденного из 10 тысяч. В Турции количество больных большое — детей с ФКУ рождается 1 на 2, 5 тысячи, в Японии – 1 на 100 тысяч рожденных, в Африке (у представителей негроидной расы) болезнь встречается исключительно редко. У девочек патология диагностируется в 2 раза чаще по сравнению с мальчиками.
В медицинской литературе можно встретить и другие обозначения болезни — фенилпировиноградная олигофрения, болезнь Феллинга.
Код по МКБ 10
Классическая фенилкетонурия в справочнике международных классификаций болезни обозначается кодом E70 (нарушения обмена ароматических аминокислот).
Лечением детей с выставленным диагнозом занимается педиатр, пациенты любого возраста также должны обследоваться и наблюдаться у психоневролога. Требуются консультации опытного диетолога, под руководством которого составляется диетотерапия.
Наследуется ли фенилкетонурия
Заболевание передается по наследству, однако только в том случае, если оба супруга (биологические родители) являются носителями дефективного гена. Но патология у детей таких пар развивается не всегда:
В 25 % на свет появляются больные малыши; В 50 % случаев дети являются носителями дефектного гена фенилкетонурии; В 25 % рождаются абсолютно здоровые по всем показателям малыши.Наличие гена ФКУ определяется в медико-генетических центрах. Если у пары уже рожден ребенок, больной фенилкетонурией, то обследование для них обязательно перед планированием повторной беременности.
Среди населения носителей патологического гена насчитывается 2-3 %, но заболевание встречается относительно редко в связи с тем, что для его развития необходимо, чтобы и у папы, и у мамы был ген, вызывающий данную болезнь.
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии — болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).
Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.
Виды
Заболевание может протекать по классическому и атипичному варианту. К атипичным формам болезни относят:
ФЕНИЛКЕТОНУРИЮ II ТИПА . Мутация гена приводит к дефициту дигидробиоптерин-редуктазы, в результате активность соединения, ответственного за преобразование фенилаланина, нарушается. Одновременно в сыворотке крови и в цереброспинальной жидкости больного выявляется дефицит витамина В9, от нормального количества которого зависит утилизация (расщепление и выведение) аминокислот. ФЕНИЛКЕТОНУРИЮ III ТИПА . Развивается вследствие нехватки катализатора, необходимого для выработки тетрагидробиоптерина (преобразует поступающий в организм фенилаланин в тирозин). ПРИМАПТЕРИНУРИЮ . Выявляется при незначительной степени гиперфенилаланинемии. До настоящего времени ферментная мутация данного вида фенилкетонурии еще не установлена. Но у больных в моче обнаруживается избыточное количество примаптерина и его соединений, а в спинномозговой жидкости уровень нейромедиаторных метаболитов остается в пределах нормы.Атипичные типы ФКУ сходны по клиническим симптомам с классическим течением патологии, но при их развитии даже своевременное лечение и диетотерапия не останавливают необратимые изменения в функционировании органов ЦНС.
Отдельно выделяют материнскую фенилкетонурию, заболевание диагностируется у детей, рожденных женщинами с ФКУ, которые не соблюдали лечебное питание.
Повышенный уровень фенилаланина вызывает ряд врожденных аномалий развития головного мозга:
Вентрикуломегалию – увеличение желудочков головного мозга в размерах; Задержку миелинизации (формирование оболочек нервных клеток) и гипоплазию (недостаточное развитие) белого вещества мозга; Низкий вес головного мозга по сравнению с нормой.Материнская фенилкетонурия становится причиной хронической интоксикации развивающегося плода токсическими соединениями, и как следствие этого приводит к задержке развития умственной функции рожденных детей.
Основные причины болезни
Основная причина развития болезни – наследование детьми от обоих биологических родителей мутантного гена. Таким образом, заболевание считается аутосомно-рецессивным.
В большинстве случаев возникает вследствие мутационных изменений гена, находящегося на длинном плече 12 хромосомы и отвечающего за кодирование фермента фенилаланин-4-гидроксилазы.
Такой вариант болезни является классической ФКУ и диагностируется у 98 больных детей из ста.
Гиперфенилаланинемия у больных при обследовании может доходить до 30 мг% и больше. Отсутствие своевременной терапии становится причиной умственной отсталости, и чаще всего она выражена значительно.
При фенилкетонурии II типа генетический дефект находится в 4-й хромосоме, прогноз неблагоприятный – смерть малыша вследствие необратимых изменений обычно происходит на 3 году жизни.
При III типе заболевания мутация находится в 11 хромосоме, эффективного лечения нет, поэтому при данном диагнозе развивается тяжелая умственная отсталость.
ВАЖНО : Близкородственные браки повышают вероятность появления детей с ФКУ.
Считается что риск рождения ребенка у пар – гетерозиготных носителей дефектного гена возрастает в следующих случаях:
При хроническом алкоголизме отца или матери; Если в период беременности у женщины и ее партнера были инфекционно-воспалительные заболевания половых органов; При негативном влиянии на организм родителей плохих экологических условий, вредной работы, хронических болезней.Патогенез
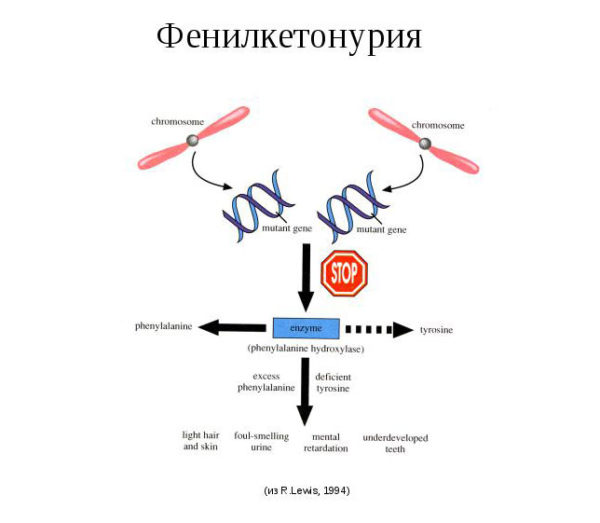
При фенилкетонурии в печени больного не продуцируется особый фермент под названием фенилаланин-4-гидроксилаза.
Основная его функция – преобразование поступающего в органы ЖКТ с пищей фенилаланина в аминокислоту тирозин. Это аминокислота входит в состав большинства ферментов, гормонов, способствует выработке меланина (пигмент), принимает участие в образовании белков и в функционировании большинства внутренних органов.
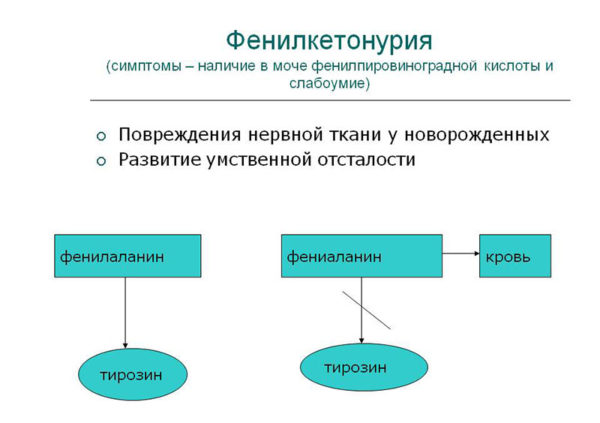
Метаболический блок при фенилкетонурии неуклонно приводит к тому, что начинают работать побочные (атипичные) пути обмена фенилаланина, в результате он трансформируется в те вещества, которых в теле здорового человека быть не должно.
Это кислоты – фенилмолочная и фенилпировиноградная, ортофенилацетат, фенилэтиламин. По сути, они являются токсическими соединениями, и их накапливание в кровеносной системе приводят:
К нарушению нормального обмена липидов (жировых клеток) в разных отделах головного мозга; К нехватке нейромедиаторов, ответственных за бесперебойную передачу нервных импульсов во всей нервной системе.В результате это приводит к прогрессирующему снижению интеллекта и к умственной отсталости выраженной степени – олигофрении, имбецильности, идиотии.
Отсутствие терапии становится причиной того, что через каждые 10 недель коэффициент развития интеллекта больного малыша падает на 5 пунктов.
Симптомы
Дети, больные фенилкетонурией, внешне на первых неделях жизни ничем не отличаются от здоровых. Поступление пищи (материнского молока или смесей) приводит к накапливанию у них в организме фенилаланина и побочных (токсических) продуктов его обмена. И обычно к 2-3 месяцем появляются первые признаки патологии, это:
Вялость; Возбудимость или наоборот заторможенность; Частые срыгивания, иногда переходящие в упорную рвоту, что может быть расценено за симптом пилоростеноза; Судороги после 3 месяцев.Из-за срыгивания и рвоты малыши не добирают в весе. Если на этом этапе начать лечение, то дальнейших патологических изменений в организме происходить не будет.
При отсутствии адекватной терапии ближе к 4-5 месяцам появляются первые признаки задержки психического развития:
Отсутствие интереса к окружающей действительности; Младенец не распознает родителей, не обращает внимания на яркие игрушки, предметы, мало реагирует на звуки; Задерживается речевое развитие.При фенилкетонурии страдает и уровень физического развития. Такие дети гораздо позже по сравнению со здоровыми сверстниками начинают активно переворачиваться, удерживать головку, сидеть.
Обратить внимание можно и на своеобразную походку – ножки широко расставлены в стороны, одновременно согнуты коленные и тазобедренные суставы. Это приводит к появлению шатающейся походки.
В позе сидя у детей скрещены и подогнуты под себя ноги и согнуты в локтевых суставах руки (поза портного). В движениях нет мягкости и плавности, нередко отмечается дрожание конечностей, гиперкинезы.
Возможно нарушение тонуса мышц, у части малышей к полутора-двум годам возникают эпилептические припадки.
При фенилкетонурии умственные и психические изменения, уже возникшие на момент начала терапии, скорректировать невозможно. Поэтому при позднем лечении у детей будет в то или иной мере наблюдаться задержка интеллектуального развития.
В тяжелых случаях к 4 годам умственные нарушения приобретают вид идиотии – самой запущенной стадии олигофрении.
Помимо интеллектуальных и психических отклонений заболевание проявляется и другой симптоматикой:
Специфическим, неприятным (некоторые называют его мышиный или плесневый) запахом от больного малыша. Характерен только для фенилкетонурии. Возникает вследствие выделения продуктов расщепления фенилаланина с мочой и через поры кожи; Изменения на кожи – появление шелушащихся пятен, развитие экземы и дерматитов. Возникают они также как следствие выхода токсических соединений через кожу; Поздно (к 15-18 месяцев) прорезываются зубы, наблюдается гипоплазия эмали; Гипопигментация. Снижение меланина становится причиной постепенного посветления кожи, волос и глаз. Обычно дети с ФКУ светлокожие с голубыми зрачками и белыми волосами, поэтому у них повышена чувствительность к солнечным лучам; Гипотонией, чрезмерной потливостью, периодическими запорами, синюшностью кожи кистей рук и стоп; Врожденными пороками сердечной мышцы.
При II типе ФКУ наблюдается сухожильная гиперрефлексия, судороги, спастический тетрапарез, тяжелая степень отклонений в интеллектуальном развитии.
Заболевание обычно быстро прогрессирует, и вследствие необратимых изменений гибель малыша происходит на третьем-четвертом году жизни.
Для ФКУ III характерно – микроцефалия, тетрапарез спастический, олигофрения.
Злокачественная форма болезни характеризуется:
Быстрым прогрессированием неврологических нарушений, в том числе и умственной отсталости (при этом уровень ФА вследствие диетотерапии может быть в пределах нормы); Нарушением глотания; Выраженным беспокойством малыша; Тяжело купирующимися судорожными приступами.При злокачественной ФКУ нервная система быстро повреждается и если вовремя не будет проведено соответствующее лечение, то смерть малыша наступит в течение нескольких месяцев.
Отказ от лечебного питания взрослых пациентов приводит к колебанию настроения, к внутреннему беспокойству, страху и депрессии , что создает социальные проблемы.
На высокий уровень фенилаланина после прекращения лечения указывает:
Бессонница; Импульсивность; Деконцентрация; Потеря способности к рациональной оценке происходящих событий.Диетотерапия необходима даже, если интеллект уже снижен. Соблюдение правильного питания позволяет снизить агрессию у больных, уменьшает чувство тревожности и улучшает контактирование с окружающими.
Психическое состояние больных ФКУ
Нелеченая фенилкетонурия приводит к глубокой степени умственной отсталости, в большинстве случаев это имбецильность или идиопатия.
Не исключается развитие эхопраксии – повторение движений за окружающими людьми, эхолалии – повторений слов. Характерна также вялость с периодическими явлениями раздражительности, агрессии и злобы.
Принципы диагностики
В настоящий момент на территории Росси массово в роддомах проводится неонатальный скрининг – определение наследственных заболеваний, в том числе и ФКУ, на 3-4 день жизни новорожденного.
Массовое обследование детей позволяет своевременно выявить фенилкетонурию и назначить соответствующее лечение, что при полном соблюдении терапии исключает дальнейшее прогрессирование патологии.
В случае отсутствия лечения (но не ранее 10 дня жизни новорожденного) в моче выявляются фенилкетоны – продукты расщепления фенилаланина.
Если в крови при проведении неонатального скрининга обнаруживаются данные за развитие патологии, то дальнейшее обследование должно проводиться на базе медико-генетических центров.
Для подтверждения диагноза необходимы следующие исследования:
Выявление уровня фенилаланина в сыворотке и высохшем пятне крови; Определение количества тирозина; Копрограмма; Потовый тест; ДНК-диагностика; Хроматография; Проба Феллинга – определение в моче фенилпировиноградной кислоты; Электроэнцефалография.В ряде случаев назначается определение показателей фенилаланингидроксилазы в биоптате печени.
Классификация ФКУ по показателю фенилаланина в сыворотке
В некоторых странах фенилкетонурия классифицируется на типы в зависимости от уровня фенилаланина в сыворотке крови:
Классическая ФКУ – показатель фенилаланина достигает 20 мг% (1200 мкмоль/л) и выше; Умеренная – от 15 до 20 % (900 -1200 ммол/л); Мягкая – от 10 до 15 мг % (600-900 ммол/л); Мягкая гиперфенилаланинемия – от 6 до 10 мг % (360-600 ммол/л); Мягкая гиперфенилаланинемия, при которой больные не нуждаются в лечении, — от 2 до 6 мг % (120-360 ммол/л).Злокачественная гиперфенилаланинемия диагностируется при недостаточности тетрагидробиоптерина.
Лечение
Единственный способ лечения на сегодняшний день фенилкетонурии – диетотерапия, при соблюдении которой из питания исключаются продукты, содержащие аминокислоту фенилаланин.
Строгое соблюдение диеты однозначно необходимо, если уровень аминокислоты в сыворотке более 2-5 мг %, если этот показатель ниже, то лечебное питание не назначается.
Важное значение имеет срок назначения лечебного питания – нервная система малышей активно продолжает развиваться на протяжении первого года жизни и именно в это время необходимо уже начать терапию.
Соблюдение диеты позволяет вырастить полноценного ребенка без отклонений в умственном и психическом здоровье. Придерживаться эллиминационной диеты многие врачи советуют вплоть до 18-20 лет, то есть до момента окончательного формирования головного мозга.
После этого возраста питание можно расширять, но под контролем уровня аминокислоты в крови. При высоких уровнях фенилаланина (что устанавливается на основании анализов) диеты необходимо придерживаться до конца жизни.
Суть диеты – полный отказ от обычных белковых продуктов, больным детям нельзя давать:
Мясо и содержащие его полуфабрикаты, рыбу; Морепродукты; Яйца; Творог и твердые сыры; Бобовые культуры; Муку из пшеницы (изделия из нее); Гречневую крупу, овсянку, манку.Диета при фенилкетонурии должна отвечать следующим требованиям:
Количество натурального белка в рацион должно быть сведено к минимуму; Белок (без фенилаланина) необходимый для нормального развития больной должен получать из специального лечебного питания; Организм обеспечивается энергией за счет использования низкобелковых продуктов; Соответствующую возрасту дозу микроэлементов и витаминов дети должны получать из дополнительных комплексов и лечебных средств для ФКУ.У малышей около 80 % потребностей организма в белке должно восполняться за счет аминокислотных смесей, в составе которых нет фенилаланина.
До года это такие марки питания, как:
Лофеналак; Нофемикс; Афенилак.После года постепенно переходят на смеси:
Нофелан; Фенилфри; Тетрафен; Бигрофен; Афенилак; МД мил ФКУ-3.
Грудное вскармливание запрещено не всегда, но обычно определяются дозы грудного молока, не приносящие вред новорожденному, в каждом случае индивидуально. Но мама при этом должна сама придерживаться строгой безбелковой диеты.
Прикорм начинают давать с овощных и фруктовых соков и пюре. Разрешены безбелковые каши – из кукурузы и риса, кисели на фруктовом соке и амилопектиновом крахмале.
После полугодия в питание можно вводить специально созданные напитки для малышей с ФКУ – Нутриген, Лопрофил, макаронные изделия, после 8 месяцев – хлеб без белков.
Для детей после года выпускаются специальные лечебные продукты:
Тетрафен; MD мил ФКУ-3; Изифен; П-АМ; ХР Максамум; ХР Максамейд.
Лечебное питание при фенилкетонурии очень дорогое, но по закону граждане России им обеспечиваются бесплатно.
Зарубежные компании выпускают специализированные малобелковые продукты – десерты, приправы, соусы, напитки, хлеб и печенье.
Маленькие пациенты с данной патологией, переведенные на лечебное питание, нуждаются в постоянном определении количества фенилаланина в сыворотке.
Первые три месяца жизни анализ необходимо сдавать еженедельно, затем до года ежемесячно, до трех лет – раз в 2-3 месяца.
При лечении необходимо добиться снижения аминокислоты до 2-6 мг, после 10 лет этот показатель должен быть не более 10 мг %.
Маленькие пациенты с фенилкетонурией находятся на диспансерном учете у детского психоневролога. Во время их осмотра контролируется:
Интеллектуальное и физическое развитие; Нутритивный статус; Эмоциональное и речевое развитие.Помимо лечебного питания больные периодически должны получать витаминно-минеральные комплексы, ноотропные препараты. При склонности к судорогам назначают антиконвульсанты – Клоназепам, Депакин. Показаны курсы массажа, физиотерапии, лечебной физкультуры.
При атипичных формах ФКУ элиминационная терапия нужных результатов не дает. Таким больным назначают гепатопротекторы, Тетрагидробиоптерин, Леводопу, противосудорожные средства. Применение этих медикаментов облегчает протекание заболевания.
Для лечения фенилкетонурии разрабатываются и уже в некоторых странах экспериментально применяются альтернативные результаты терапии:
Заместительная терапия фенилаланинлиазой – это растительный фермент, способствующий расщеплению аминокислоты до нетоксичных для организма соединений; Генетическая инженерия – введение больным синтетически воспроизведенного нормального гена взамен дефектного; Методика под названием «большие нейтральные аминокислоты» — использование препаратов, снижающих всасываемость поступающего с белковой пищей фенилаланина и ограничивающих его попадание в головной мозг.
Диетотерапия при классической форме болезни по мнению большинства врачей должна соблюдаться на протяжении всей жизни. При мягких формах заболевания возможно постепенное расширение питания.
Но девушки с ФКУ должны знать, что в репродуктивном возрасте им важно придерживаться элиминационной диеты как до наступления зачатия, так и на всех триместрах беременности.
Если диеты не придерживаться, то младенец рождается с множественными пороками развития:
У 92 % новорожденных диагностируется умственная отсталость; У 73 % малышей микроцефалия; У 12 % диагностируются пороки сердца; У 40 % — выявляется низкая масса тела на момент рождения.Чтобы ребенок родился от больной ФКУ женщины здоровым необходимо как минимум за 2 месяца до зачатия перейти на низкобелковую диету и придерживаться лечебного питания все 9 месяцев.
При этом нужно постоянно контролировать уровень фенилаланина, он должен быть не выше 4 мг %.
Прогноз и профилактика
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.